
Dr. Joanne Matsubara, PhD.
Professor, Department of Ophthalmology & Visual Sciences, Director of Research, Basic Sciences, Eye Care Centre, Assistant Director, Centre for Macular Research
The Matsubara Lab
For directions to lab, see link. Or visit our lab website.
Biography
Associations
Member of Brain Research Centre
Member of Graduate Program in Neurosciences
Member of Graduate Program in Cell and Developmental Biology
Associate Member of Cellular and Physiological Sciences
Ph.D.University of California, San Diego (USA)
AMD is a multifactorial, degenerative eye disease and a major cause of blindness among the elderly worldwide. Late stage, dry form of AMD (geographic atrophy, GA), is characterized by high drusen load with slow deterioration of the RPE and subsequent loss of the photoreceptor cell, resulting in irreversible blindness. Currently, antioxidant vitamins and zinc supplements are suggested to slow disease progression, but do not stop vision loss, and does not work for all patients with dry AMD. Our recent work suggests that RPE loss in GA is induced by the activation of the inflammasome, a cytoplasmic receptor complex that senses intracellular stress signals and causes pyroptosis, a caspase 1-dependent form of cell death.
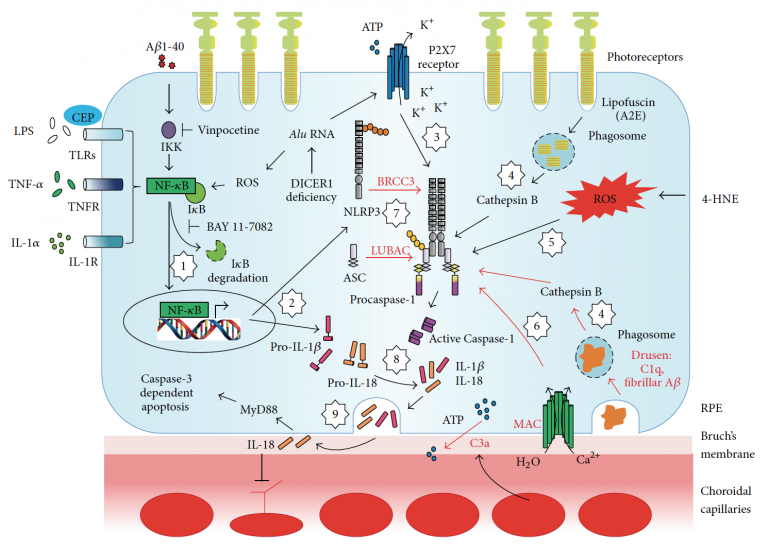
This project will focus on a gene treatment to bolster levels of X-chromosome linked inhibitor of apoptosis protein (XIAP), an experimental neuroprotective agent for photoreceptors in retinal dystrophies. A recent important finding is that XIAP, well known for its ability to suppress caspase-3 dependent apoptosis, regulates and inhibits inflammasome activity. We propose to bolster protein levels of XIAP by gene therapy in vitro and in vivo to identify the dual mechanisms of XIAP rescue of RPE. The broad goal of this research project is to prevent blindness in age-related macular degeneration (AMD) by development of a novel method to limit inflammation-induced atrophy of the retinal pigment epithelium (RPE) in the eye.
AMD is a complex, multifactorial disease and a major cause of blindness among the elderly worldwide. The more common ‘dry’ form accounts for over 90% of AMD, and is characterized by drusen, extracellular deposits of unwanted cell debris located near the RPE. Drusen become larger and more numerous during dry AMD progression, culminating in geographic atrophy (GA), in which RPE and photoreceptors die, resulting in blindness. Genetic factors play an important role in AMD development. Namely, complement factor H gene variants are strongly associated with increased risk of AMD.
Preliminary gene and protein studies suggest that RPE genotyped with the CFH risk variant demonstrate reduced levels of (inhibitory) complement regulators that promote cellular deposition of membrane attack complex (MAC, complement cascade end-product). Here, we test the role of MAC on inflammasome activation, a cytoplasmic receptor complex that is deemed to play a role in chronic inflammation and AMD development. How MAC alone, or in combination with drusen components (e.g. amyloid beta), promotes inflammasome activation in RPE is unknown. Results will inform on the genotype-specificity of the cellular mechanisms associated with the pro-inflammatory cascades that contribute to RPE dysfunction associated with GA in AMD.
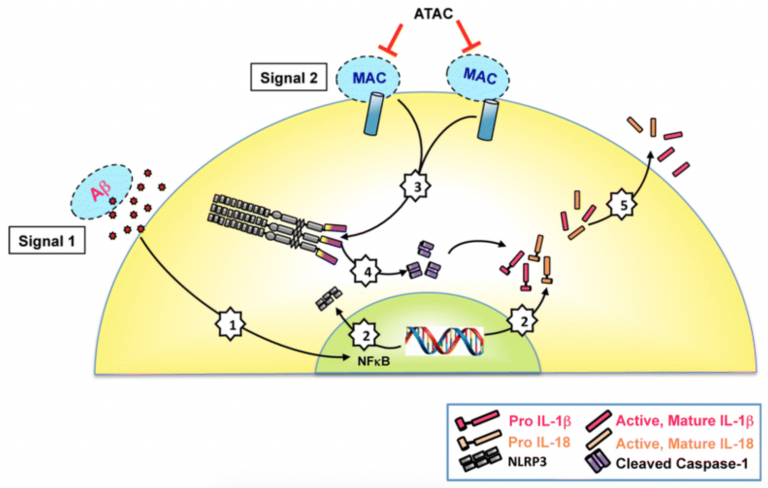
The goal of our work is to prevent chronic inflammation, an important initiating trigger in age-related macular degeneration (AMD) pathogenesis. To achieve this goal, we will identify the cellular interactions between the complement system and inflammasome activation in retinal pigment epithelial (RPE) cells genotyped for complement factor H (CFH) gene variants.
Age-related macular degeneration (AMD) is the leading cause of irreversible blindness in the elderly. Unfortunately, current treatments only exist for the advanced form of AMD and begin after significant eye damage has already occurred. Chronic inflammation and abnormal formation of new blood vessels play a significant role in this complex multifactorial disease. Our proposal will focus on special enzymes, called proteases that shape the cellular environment in the retina. Abnormal activation of these proteases can cause inflammation by releasing harmful molecules such as tumor necrosis factor (TNF) in the eye. Our group has developed a way to use the proteases so that instead of harmful factors, they release therapeutic factors. It is important that the therapeutic factor is only released when it is needed to minimize any potential side effects in the retina The long term goal of this novel and first-in-class strategy is to have the retinal cells release the engineered therapeutic factors only at the site of inflammation and injury, in timely manner, and only when the injurious processes have reached a certain critical threshold. Our proposed "smart" therapeutic strategy will be positioned in retinal cells in advance of disease, and work as needed to reduce the negative systemic and off-target effects frequently associated with current therapies for eye disease.
Traumatic eye injury is one of the leading causes of monocular blindness worldwide. This profound and frequently irreversible posttraumatic loss of vision has a poor prognosis due to retinal cell death, scar formation, and lack of functional regeneration. Proliferative vitreoretinopathy (PVR), a form of intraocular fibrosis, is often the primary reason for the loss of vision after ocular trauma, and frequently occurs after blunt trauma and open globe injuries caused by penetration, rupture, perforation, and presence of intraocular foreign bodies as well as after retinal re-attachment surgery. PVR is a complex cellular process initiated by inflammation, ischemia, and hemorrhage within the vitreous cavity. These events trigger the migration, and proliferation of inflammatory cells in the retina, including the retinal pigment epithelium and glia which then contribute to the formation of epiretinal membranes which contract and separate the retina from the RPE thus causing retinal tears, degeneration and gliosis with disastrous consequences for vision. Many of these pathological events can be traced to the local pro-inflammatory processes that trigger several downstream mechanisms including the activation of metalloproteinases (MMP), that facilitate extracellular matrix (ECM) remodeling. The inflammation-induced MMP have also been shown to participate in the shedding of various transmembrane proteins in the CNS including receptors, adhesion molecules, growth factors, and cytokines.
Our group will use an innovative strategy to genetically engineer transmembrane proteins ready to release their anti-inflammatory, anti-proliferative and neuroprotective ectodomains by extracellular proteases activated under adverse conditions induced by traumatic eye injury. We hypothesize that, once inserted into the cell membrane of retinal cells, these “protease activity sensors” will be activated by traumatic injury-induced inflammatory events to release “therapeutic ectodomains”. We propose here to exploit the released ectodomains to avert the pathological cascade that leads to irreversible loss of vision in PVR.
One of the biological hallmarks of Alzheimer's Disease (AD) is the abnormal deposit of amyloid beta in brain tissues, and recently we have identified that this also occurs in the retina. This study will utilize transgenic mouse models of AD to investigate the relationship of amyloid beta buildup in the brain and eye. By quantifying the ratio of amyloid beta plaques in the brain with deposits in the eye at different ages, we will obtain important information to estimate the relative plaque load in the brain based on the measurements obtained in the eye.
Next, preclinical studies of post-mortem eye tissues from AD, non-AD dementias, and age matched control donors will be undertaken in the whole mount preparation and will provide us with measurements of amyloid beta deposits in the AD eye, and its relationship with amyloid beta plaque load in the brain.
We will use the endogenous fluorescence of curcumin, a natural and safe fluorochrome that has a high affinity to a amyloid beta plaques, to label amyloid beta deposits in the eye towards the development of specialized fluorescent imaging equipment for ocular amyloid beta deposits. Our non-invasive optical imaging technology will continue to undergo development towards improving our understanding of the disease mechanisms and accelerate the development of therapeutics. The optical imaging and computational anatomy technology tools developed here can additionally be used to investigate the role of amyloid beta in other important age related diseases (such as age related macular degeneration, AMD, and glaucoma), as well as for the development of additional biomarkers for future studies for AD.
For a complete list of our publications, please refer to PubMed.
UBC Website(s)
To learn more about the diseases we study and the research programs/networks we are in, click here.